
Craniossinostose
O QUE É?
A craniossinostose – também conhecida como craniostenose – é uma anormalidade congênita, isto é, que se apresenta desde o nascimento, podendo atingir 1 em cada 2.500 nascidos.
Essa malformação causa alterações no formato do crânio e pode levar à hipertensão intracraniana. Da mesma maneira que o aumento da pressão arterial pode trazer problemas para a saúde do indivíduo, a hipertensão intracraniana também provoca consequências como dor de cabeça, alteração visual e prejuízo na capacidade de aprendizado.
POR QUE OCORRE?
A craniossinostose é provocada pelo fechamento precoce de suturas cranianas, responsáveis pelo crescimento dos ossos do crânio.
As causas são diversas, podendo ocorrer por força mecânica (ambiente uterino), distúrbios metabólicos e, na maioria das vezes, por alterações genéticas, o que não implica necessariamente ter alguém na família com a mesma característica.
CLASSIFICAÇÃO
A maioria dos casos de craniossinostose se manifesta na forma isolada e, quando associada a outras características, pode representar síndromes genéticas, o que corresponde a 15% dos casos.
Existem quatro tipos principais de craniossinostoses na forma isolada, dependendo da sutura craniana afetada. São elas: escafocefalia, plagiocefalia anterior, trigonocefalia e braquicefalia.
Já a forma sindrômica é aquela em que a criança nasce com craniossinostose associada a outras características. As síndromes de Apert, Crouzon, Muenke, Pfeiffer e Saethre-Chotzen são os exemplos mais comuns de síndromes genéticas associadas à craniossinostose.
Essa condição congênita, portanto, pode trazer diferentes tipos de comprometimentos aos indivíduos, conforme cada caso.

COMO É O TRATAMENTO?
Assim como acontece com outras condições tratadas no HRAC-USP, o processo de reabilitação de pacientes com craniossinostose também envolve a atuação de uma equipe multidisciplinar, neste caso, composta por profissionais da área médica (cirurgiões craniofaciais, neurocirurgiões e anestesiologistas), da Odontologia, da Fonoaudiologia, da Enfermagem, da Genética, da Psicologia e do Serviço Social.
Após serem avaliados pelos profissionais das diferentes especialidades, os pacientes fazem vários exames, como tomografia de crânio e ressonância magnética (para detectar as alterações craniofaciais), monitorização não invasiva por sensor (para avaliação da pressão intracraniana) e retinografia (para avaliação do fundo do olho), entre outros.
Existem várias causas genéticas bem definidas relacionadas especialmente às craniossinostoses sindrômicas, que podem ser identificadas por meio de exames específicos de DNA. Entretanto, geralmente, o diagnóstico é clínico, isto é, realizado a partir da avaliação do conjunto das características apresentadas pela criança. Os profissionais de Genética do HRAC-USP avaliam o paciente junto à Equipe de Cirurgia Craniofacial, com o objetivo de definir o diagnóstico e, a partir daí, informar os pais sobre a causa e realizar adequado aconselhamento genético, orientando o casal sobre a chance de outros filhos terem as mesmas características.
A Psicologia, por sua vez, atua junto aos familiares, auxiliando na elaboração do diagnóstico e das informações acerca do tratamento, mobilizando seus recursos de enfrentamento para lidar com a situação. Também realiza avaliação sistematizada do Desenvolvimento Neuropsicomotor (DNPM) da criança, pois o quadro de craniossinostose pode ocasionar atrasos nesse desenvolvimento. Assim, no decorrer do tratamento, é necessário acompanhar as funções cognitivas da criança para minimizar possíveis impactos em sua vida cotidiana.
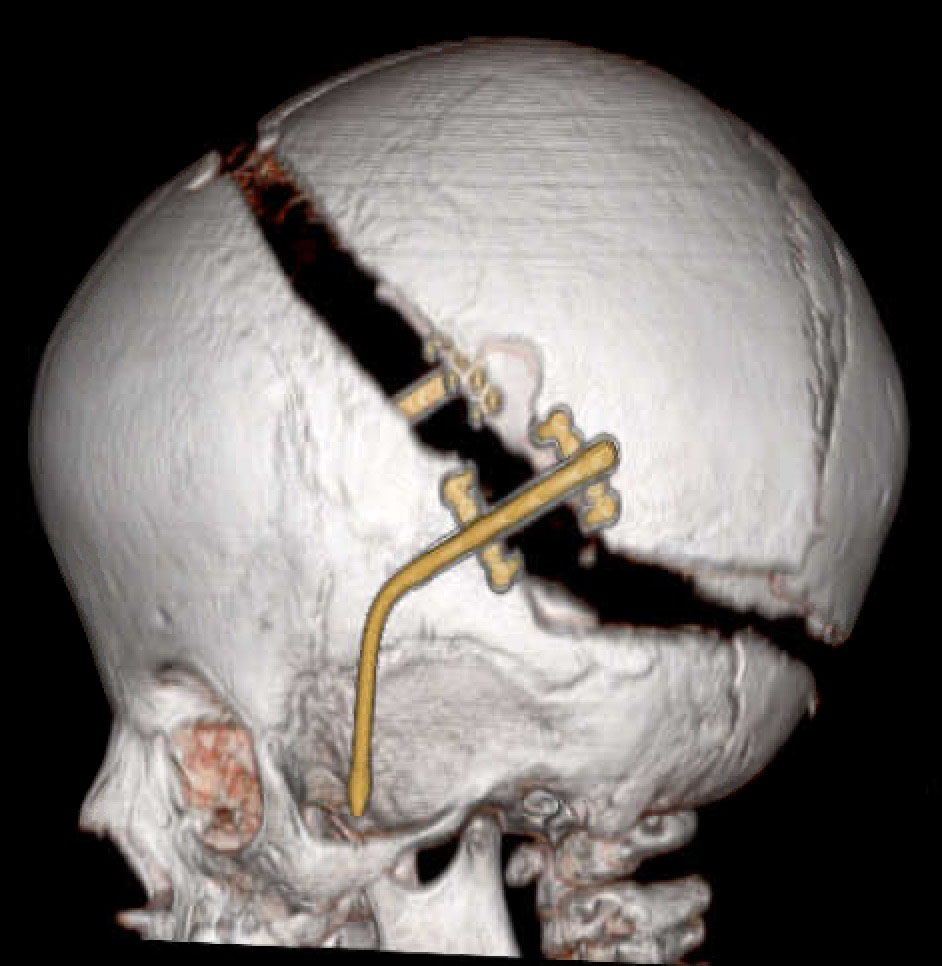
Quando o diagnóstico de craniossinostose é confirmado, vem a etapa do preparo para o tratamento cirúrgico. O mais importante é quando fazer a cirurgia! O ideal é a realizar a cirurgia durante o primeiro ano de vida (e, dependendo do diagnóstico, até os 6 meses de vida). A cirurgia para tratamento de craniossinostose no HRAC-USP é realizada em conjunto por especialistas em cirurgia craniomaxilofacial e em neurocirurgia e visa remodelar o crânio, prevenindo a hipertensão intracraniana (especialmente nos mais novos), melhorando a respiração e problemas oculares (especialmente nos sindrômicos). Independentemente da técnica empregada, o fundamental é fazer a cirurgia na idade certa, especialmente a descompressão do crânio, que deve ser realizada quando a criança ainda tem meses, antes de completar o primeiro ano de vida.
Para aumentar a segurança do paciente durante a cirurgia é necessário contar com médicos anestesiologistas com experiência neste tipo de cirurgia e que acompanham a criança ou o adulto desde o preparo pré-operatório, durante toda a cirurgia e também no pós-operatório imediato. Sua atuação envolve a administração da anestesia, o manejo de dor e sangramento e, eventualmente, o controle avançado de via aérea, pois a craniossinostose pode causar dificuldade na intubação.
O cuidado de Enfermagem ao paciente com craniossinostose também demanda conhecimento técnico-científico específico. As intervenções ocorrem desde o pré-operatório até o preparo para a alta hospitalar, devendo estar centradas na família, com foco no autocuidado, fomentando a identificação precoce de intercorrências e, consequentemente, a prevenção de complicações e a plena recuperação.
As repercussões faciais podem levar a alterações odontológicas (tanto nas craniossinostoses sindrômicas como nas isoladas), como por exemplo: desalinhamento dos dentes; falta de encaixe entre as arcadas dentárias; ausência de alguns dentes; falta de espaço para todos os dentes irromperem (nascerem) na boca etc. O acompanhamento odontológico deve ser feito já nos primeiros anos de vida, com um Odontopediatra – quando os dentes de leite iniciam o seu aparecimento na boca (entre 6 meses e 2 anos de vida) –, por meio de cuidados iniciais como prevenção de cáries. A partir dos 6 anos, quando tem início o nascimento dos dentes permanentes, o paciente já pode realizar uma avaliação com o Ortodontista, através do monitoramento das trocas dentárias (substituição dos dentes de leite pelos dentes permanentes) e, se necessário, uso de aparelhos ortodônticos em casos específicos para favorecer o melhor alinhamento dentário.
As craniossinostoses isoladas (não sindrômicas), de um modo geral, não costumam estar associadas a problemas de comunicação ou outros problemas funcionais, desde que o tratamento seja realizado da maneira adequada. Por outro lado, as craniossinostoses sindrômicas caracterizam quadros mais complexos e podem estar relacionadas a diversos problemas funcionais, incluindo os distúrbios de fala, linguagem, audição e alimentação. O acompanhamento fonoaudiológico desde o nascimento até a idade adulta é sempre indicado nessas condições, e envolve o gerenciamento da alimentação, da audição, do desenvolvimento da linguagem, dos aspectos oromiofuncionais e de aprendizagem, dentre outros.
Já os profissionais do Serviço Social atuam com o objetivo principal de viabilizar o acesso e a continuidade do tratamento dos pacientes. Dão orientações sobre direitos sociais, articulam recursos de apoio ao tratamento, prestam atendimento social e acompanham situações familiares e sociais complexas, decorrentes das anomalias craniofaciais. Além disso, buscam realizar intervenções junto aos recursos da comunidade (Tratamento Fora de Domicílio-TFD, Prefeituras, Secretarias), visando facilitar a longa permanência do paciente em Bauru, principalmente nos casos cirúrgicos mais complexos.
É importante destacar que o tratamento integral envolvendo todas essas especialidades é de fundamental importância para que sejam alcançados os melhores resultados ao longo do tratamento.
COMO ENCAMINHAR UM PACIENTE?
A assistência prestada no HRAC-USP é integralmente via Sistema Único de Saúde (SUS). O acesso de novos pacientes com suspeita ou diagnóstico confirmado de craniossinostose é por meio das Centrais de Regulação do SUS. O agendamento, portanto, não é feito pelo HRAC-USP. A família deve procurar, primeiramente, a rede básica de saúde do seu município, para avaliação inicial e posterior encaminhamento via Sistema Informatizado de Regulação do Estado de São Paulo (SIRESP) – conhecido anteriormente como CROSS. O programa de Anomalias Craniofaciais do HRAC-USP (no qual são atendidos os pacientes com craniossinostose) tem regulação nacional e, portanto, recebe pacientes de todos os Estado do Brasil.
(Fonte: Equipe multidisciplinar de Cirurgia Craniofacial do HRAC-USP)
CONTATO (para dúvidas, orientações e mais informações)
Serviço de Prontuário de Pacientes • HRAC/Centrinho-USP (caso novo)
Horário de atendimento: de segunda a sexta-feira, das 8h às 17h
e-mail: casonovohrac@usp.br
telefone: (14) 3235-8128